
Molecular Docking Studies of Potential Drugs for Covid19 Targetting on the Coronavirus Hemagglutinin Esterase
DOI:
https://doi.org/10.37934/jrnn.4.1.1318Keywords:
Covid19, hemagglutinin esterase, molecular dockingAbstract
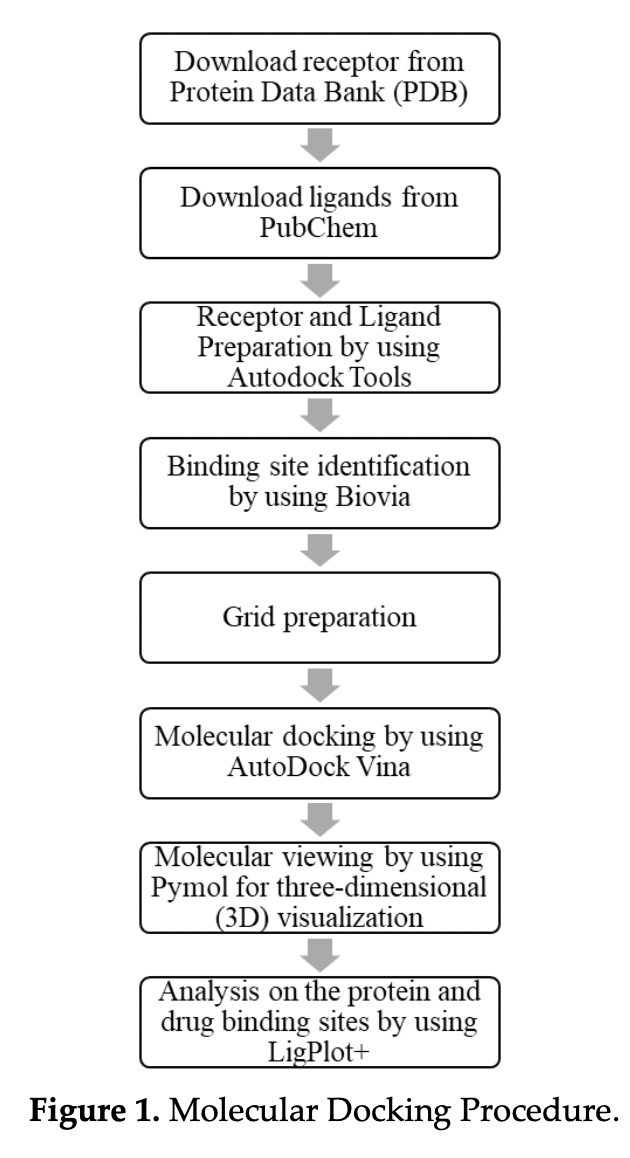
The goal of this project is to contribute, using molecular docking simulation, to the search for potential drug candidates for Covid-19. The COVID19 receptor used in this study was coronavirus hemagglutinin esterase and the drugs were spirosolane, oridonin and silymarin. The protein and the ligands were downloaded from the protein data bank (PDB) and PubChem website, respectively. Using Autodock Tools, all downloaded proteins and ligands were then prepared. AutoDock Vina was used to perform molecular docking. The best binding sites were selected based on the ranking of binding energy or binding affinity given in kcal/mol. It was found that all three ligands produced low binding energies between -8 to -10 Kcal/mol. The analysis on molecular interactions were carried out to investigate the formation of hydrogen bonds and hydrophobic interactions in all docked conformations and silymarin was found to be the best ligand out of the three in terms of binding to the coronavirus hemagglutinin esterase.
Downloads
Downloads
Published
How to Cite
Issue
Section













